5 updates to look for in the latest ICH GCP E6 guideline (R3)
The International Council of Harmonization (ICH) published earlier this year its long anticipated draft guideline ICH E6 (R3) on Good Clinical Practice (GCP) which is currently open for public consultation. Regulatory agencies around the globe gather comments from the public which will then be transferred to ICH for revisions on the draft document. While for some agencies this deadline is already reached, for others there is still some time left, including the EMA (Europe), HSA (Singapore), Health Canada (Canada) and Swissmedic (Switzerland). So, if you are planning to leave a comment or if you are just interested on this topic, here is our overview of some of the most interesting new features included in the new draft that will create lots of discussions but also shape the landscape of the clinical space of the future.
Data governance, computer systems, digital solutions as well as their providers are arguably topics which regulators needed to address to streamline their expectations when it comes to compliance within clinical trials. This was evident with the latest update in the previous guidance document, ICH E6 (R2) as well as in various agency-specific guidance documents such as the recently finalized EMA guideline on computerized systems and electronic data in clinical trials and the draft FDA guidance for industry on Electronic systems, Electronic Records, and Electronic Signatures in Clinical Investigations Questions and Answers .
How are these topics addressed in the ICH E6 ( R3)? Here is our take on our favorite 5:
1. Quality by Design (QbD)
QbD is included as a term within the ICH guideline Q8 (R2) on pharmaceutical development and refers to the “systematic approach to development that begins with predefined objectives and emphasizes product and process understanding and process control, based on sound science and quality risk management”. In the draft ICH E6 (R3), QbD is now included within the ICH GCP principles with the goal of identifying:
- critical factors that ensure trial quality
- risks that threaten the integrity of the critical factors and the reliability of the trial results
What is expected in other words, is to design clinical trials and their components while taking into consideration the factors which are important for ensuring that trial participants are protected and that trial results are reliable and can be interpreted in a way that allows for regulatory decision making. When it comes to data considerations, efforts could concentrate on holistic approaches that use data governance and knowledge management to ensure data integrity, as thoroughly described within the GAMP RDI Good Practice Guide: Data Integrity by Design.
2. Data Lifecycle
As data is detrimental for ensuring trial results’ reliability, the current revision asks sponsors to ensure that processes are documented and implemented in a way that preserves data integrity throughout the data lifecycle. Such procedures should focus mainly on the following areas and are applicable for both electronic and paper records as well as systems developed or operated by external suppliers:
- Data capture
- Metadata and audit trails
- Review of data and metadata
- Data corrections
- Data transfer, exchange and migration
- Dataset finalization prior to analysis
The principle follows the data lifecycle approach (Figure 1) as this is described within the ISPE GAMP Guide: Records & Data Integrity and is adapted to address the clinical trial setting. Specific attention is given to data handling, where sponsors are expected to ensure data integrity and confidentiality by applying quality control and quality assurance throughout the data lifecycle following a risk-based approach or in other words focusing on data based on their criticality and impact on patient safety and trial quality.
Investigators are also expected as part of their responsibilities, to contribute to the integrity of the data when it comes to reporting, verifying and interpreting clinical trial data.
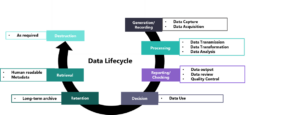
Figure 1: Clinical data lifecycle, adapted from Data Integrity in Global Clinical Trials: Discussions from Joint FDA and MHRA GCP Workshop, doi:10.1002/cpt.1794
3. Supplier management
While in the last revision (R2) the focus was exclusively on Contract Research Organizations (CROs), here in (R3) the respective CRO section is not included and the term “service providers” appears within the “Sponsor” chapter instead. To our understanding, this change addresses the current clinical trial landscape that involves multiple vendors for the management of trials, including those that develop or manage digital solutions or computerized systems for data capture, document management, statistical analysis, Investigational Product (IP) management, IP blinding and many others.
Regarding supplier management, agreements should be made between sponsors and 3rd parties, documented prior to a trial, and updated when significant changes take place. These agreements need to include any supplier-undertaken activities although the ultimate responsibility for these activities remains with the sponsor. A new addition focuses on the fact that service providers that undertake clinical trial activities need to implement appropriate quality management including incident reporting to ensure patient safety and result reliability.
These new points will eventually help the focus points during supplier auditing when it comes to vendor qualification by the sponsors, as the regulators now express their expectation that sponsors have direct access to service providers’ quality documentation and that the latter operate in compliance with GCP principles.
4. Trial- specific validation of Computerized Systems
This is a topic which was initially mentioned within the recently finalized EMA guidance and has since then been the initiator of heated discussions when it comes to validation responsibilities. In the current draft, it is clearly stated that systems, as well as their interfaces need to be validated not only for their functionality but also for protocol specific configurations.
What does this mean in practice? Systems which have been validated for a certain trial can not be used in subsequent trials by the same sponsor without additional validation. Further validation is required to address trial-specific features that might impact the generated data.
We reason that since the general recommendation is to follow a risk-based approach for the conduct of clinical trials, this point here is also open for interpretation. Could a sponsor still use a system without a new round of validation if it is appropriately documented and justified why previous validation efforts are still applicable especially in cases that no new functionalities are used? Early engagement with the authorities and appropriate documentation would be helpful here.
5. Assessment of investigator/institution-deployed systems
This new addition within the data handling section of the sponsor-dedicated chapter, asks sponsors to evaluate the electronic systems which are used within investigator sites. This means that sponsors should assess if the systems used for various hospital activities such as electronic health records or record keeping are fit for the purpose of a given trial before the selection of a given investigator site.
Although this requirement makes perfect sense when it comes to maintaining data integrity throughout the data life cycle by focusing here on data generation, it also allows for a number of questions:
- How are these assessments going to be conducted? Will investigator sites accept sponsors auditing their systems or it would be sufficient to provide evidence, based on investigator interviews?
- Will this mean that only establishments that support fit-for-purpose infrastructure can from now on participate in clinical trials, therefore limiting the number of potentially available investigator sites?
- How is fitness-for-purpose defined here? Does a system need to be evaluated once or each time before the beginning of a certain trial?
All these considerations might need to be addressed during the finalization of the document to enable parties preserve data integrity within the trial lifecycle while at the same time allowing a pragmatic approach when it comes to clinical trial design and conduct.
If you have any questions on the topics discussed here or you need support in implementing these requirements, do not hesitate to contact us.