EU-GMP Chapter 4 Revision: Anticipated Changes in Documentation and Data Governance
Regulatory frameworks may be comprehensive, but documentation continues to present a consistent compliance challenge. The European Medicines Agency (EMA) reported that 45% of GCP inspection findings were related to documentation deficiencies1, a clear reminder that accurate and timely recordkeeping remains a critical area for improvement.
To address these ongoing issues, the European Commission has released the 2025 draft revision of EU-GMP Chapter 4 (Documentation) for public consultation2. This marks the most significant revision to this chapter in over a decade (Figure 1), aiming to harmonize with global standards, close gaps in outsourcing practices, and reflect advances in manufacturing technologies alongside the growing influence of digitalization3.
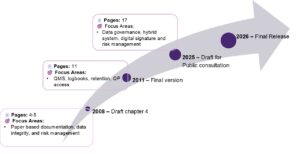 Figure 1: Evolution of Chapter 4 documentation from 2011 guideline to 2025 draft for public consultation.
Figure 1: Evolution of Chapter 4 documentation from 2011 guideline to 2025 draft for public consultation.
At a recent webinar4, Abubakar Muhammad, a compliance consultant in GxP-CC, opened the discussion with a fundamental question: “What is documentation?” The answer was: “Documentation is the living backbone of compliance, traceability, and product quality”.
During the webinar, we asked participants how they viewed the expansion of clauses from 32 in the 2011 version to 85 in the 2025 draft on documentation. 67% responded that they preferred more detailed guidance from the regulation (Figure 2 A). This result aligns closely with the latest draft, where the number of clauses has nearly tripled (Figure 2 B). The expansion reflects the regulators’ intent to provide greater clarity and to address critical topics such as data governance systems, data integrity, and lifecycle management in much more depth. In practice, more detailed guidance means fewer ambiguities, stronger alignment with modern practices, and a more robust foundation for ensuring product quality and patient safety.
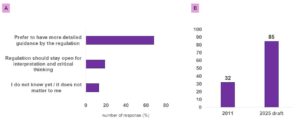 Figure 2: Panel A: Quantitative increase in the number of clauses from the 2011 version to the 2025 draft Panel B: displays results from a live webinar poll in which participants responded to the prompt: “The number of clauses in the new draft Chapter 4 more than doubled. What do you think about it?”
Figure 2: Panel A: Quantitative increase in the number of clauses from the 2011 version to the 2025 draft Panel B: displays results from a live webinar poll in which participants responded to the prompt: “The number of clauses in the new draft Chapter 4 more than doubled. What do you think about it?”
As Ulrich Köllisch, a partner in GxP-highlighted in our webinar, the language of the draft makes this clear: in the 2008 draft, the word risk appeared once; in the 2025 draft, it is mentioned 54 times. References for Data increased from 17 mentions to 157, and ALCOA, previously absent, now appears 4 times. This sharp increase signals a clear shift in the pharma industry toward risk-based thinking, robust data governance systems, and a stronger emphasis on data integrity.
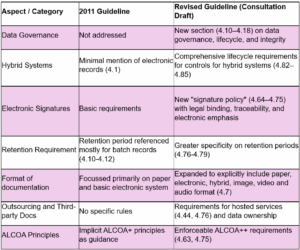
To better understand the scope of change, the table below contrasts the 2011 EU-GMP Chapter 4 guidelines with the 2025 EMA draft update. It highlights how the framework has evolved from traditional paper-based recordkeeping toward a modern, lifecycle driven approach that incorporates digital tools, data governance and risk management (Table 1).
Table 1: Comparative analysis of EU-GMP Chapter 4 (Documentation), highlighting the key differences between the 2011 guidelines and the 2025 draft update
The Big New Arrival: Data Governance
One of the most significant additions in the 2025 draft is the dedicated section on data governance. It’s more than a compliance update; it’s a shift in mindset After going through the data governance section, Ulrich Köllisch mentioned that the revision requires pharmaceutical companies to implement a robust data governance system integrated into their quality management framework, regardless of data format. This system should manage data integrity across the entire lifecycle using a risk-based approach. In practice, the system should:
- Cover the entire data lifecycle from creation and processing through verification, decision-making, secure retention, and controlled destruction
- Ensure accuracy, completeness, traceability, and protection against unauthorized changes or loss.
- Assess both data criticality (impact on product quality and decisions) and data risk (likelihood of alteration or deletion), supported by strong systems, technologies, security measures, and expertise.
- Define data accountability, including data ownership, ongoing risk mitigation and review, and communication of any residual risks to management.
The draft revision of Chapter 4 also refers to emerging guidance such as the proposed Annex 22 on AI and digitalization5. Regulated users are accountable for data integrity, and electronic records must not be converted to paper unless validated or compliant with hybrid system rules.
Data Retention
Each record should be clearly associated with its respective activity and location, regardless of the technology or service used. Risk-based control measures must be in place to safeguard record integrity throughout its lifecycle. Additionally, for an investigational medicinal product, batch documentation must be retained for a minimum of five years following the completion of the last clinical trial. The data retention strategy should consider the retention period, system retirement, security, availability, access control, and the contributed readability and accessibility of the record over time.
ALCOA ++
One of the most notable updates in the new EU GMP Chapter 4 draft is the expansion of the well-known ALCOA principles for data integrity. Originally standing for Attributable, Legible, Contemporaneous, Original, and Accurate, the framework was previously extended to ALCOA+ with the addition of concepts such as completeness, consistency, and enduring. The latest draft now introduces another “plus,” evolving the framework into ALCOA++. The new addition is Traceability, defined as the ability to trace the history, modification, or location of data through recorded identifications. This strengthens the expectation that data should not only be reliable and accurate but also fully trackable across its lifecycle.
In conclusion, the webinar turned out to be a highly interactive and insightful session. Our GxP-CC team not only delivered clear explanations but also sparked lively discussion among the participants. The audience engaged actively on key topics such as ALCOA++, traceability and major changes outlined in the consultation draft. If the 2025 Chapter revision 4 draft is approved and implemented, the team at GxP-CC would be happy to support pharmaceutical companies in several ways.
- Assist with gap assessments by comparing existing SOPs with the draft requirements to enhance operational efficiency.
- Guide the implementation of a robust data governance framework to ensure compliance and strengthen data integrity across your organization.
- Evaluate AI-based tools digital solutions by providing tailored training and awareness programs for your team.
Not sure where to begin? Connect with GxP-CC to reinforce your compliance practices, protect both patients and your business, and support healthcare innovation built on transparency and reliable data.
References
- Sellers, J. W., Mihaescu, C. M., Ayalew, K., Kronstein, P. D., Yu, B., Ning, Y. M., … & Khin, N. A. (2022). Descriptive analysis of good clinical practice inspection findings from US Food and Drug Administration and European Medicines Agency. Therapeutic Innovation & Regulatory Science, 56(5), 753-764. Descriptive Analysis of Good Clinical Practice Inspection Findings from U.S. Food and Drug Administration and European Medicines Agency – PMC
- European Medicines Agency. EudraLex Volume 4: Good Manufacturing Practice Guidelines – Chapter 4 (Documentation) [Draft for public consultation]. 7 July 2025. Available from: https://health.ec.europa.eu/consultations/stakeholders-consultation-eudralex-volume-4-good-manufacturing-practice-guidelines-chapter-4-annex_en
- European Commission. EudraLex Volume 4: Good Manufacturing Practice Guidelines – Chapter 4 (Documentation). 30 June 2011. Available from: https://health.ec.europa.eu/document/download/104b3eb8-81a7-4858-9419-cb06562adb66_en?filename=chapter4_01-2011_en.pdf
- GxP-CC. Episode 1: Mastering the 2025 EU-GMP Chapter 4 Draft Update [webinar]. 2025 Aug 7. Presented by Dr. Ulrich Köllisch, Abubakar Muhammad & James R. Francum. Available from: https://www.gxp-cc.com/insights/webinar/mastering-the-2025-eu-gmp-chapter-4-draft-update/
- European Medicines Agency. EudraLex Volume 4: Good Manufacturing Practice Guidelines – Annex 22 (Artificial Intelligence) [Draft for public consultation]. 7 July 2025. Available from: https://health.ec.europa.eu/consultations/stakeholders-consultation-eudralex-volume-4-good-manufacturing-practice-guidelines-chapter-4-annex_en