Data Integrity Challenges in the Scope of Clinical Trials – Part 2
In this 2nd part of the article series, we discuss data integrity challenges and the main outcomes in respect to eSource and EHRs in context with current regulations and expert views.
Data integrity challenges in eSource Data
With the adoption of new technologies in clinical trials, source data can be collected and managed via various means that do not always include the investigator sites. eSystems, electronic Clinical Outcome Assessments (eCOAs), electronic Patient Reported Outcomes (ePROs), e-health records (EHRs), and Interactive Response Technologies (IRT), are all different ways to capture source data in clinical trials. To safeguard proper data integrity and cope with data integrity challenges, their adoption requires a robust Quality Management System (QMS). When using eSource data, it is often the case that the data is collected and managed by third-party vendors such as central labs or technology providers, however, it always lies under the investigator’s responsibility. Therefore, the sponsors should ensure that the source data or certified copies are always be provided to the investigator, regardless of the collection method.
Lack of investigator access to source data is one of the most frequently noted BIMO (FDA’s Bioresearch Monitoring Compliance Program) deficiency. Noted as a finding during FDA inspections, it involves both the investigator for failing to maintain complete records as per 21CFR 312.62 as well as the sponsor for failing to provide the clinical investigator with information necessary to conduct the investigation properly as per 21 CRF 312.50.
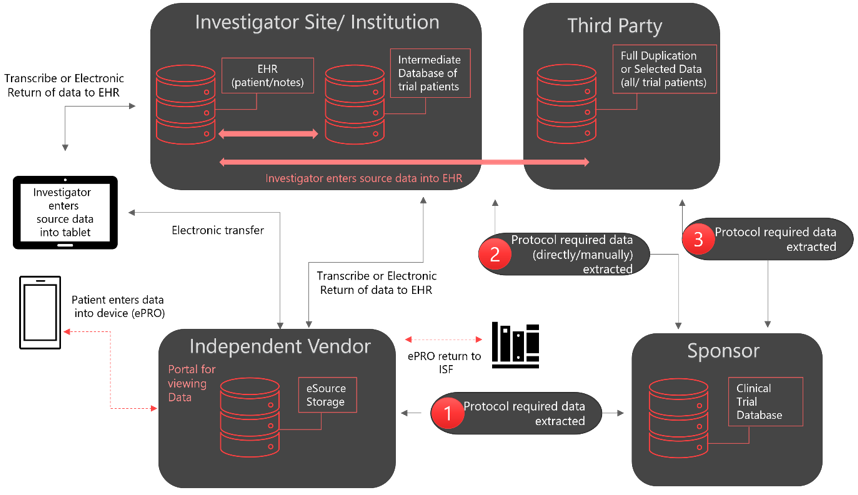
Figure 1: eSource direct Capture Models. The arrows indicate the data flow and dotted lines, possible data flow. EHR; Electronic Health Records, ePRO; electronic Patient-Reported Outcome, ISF; Investigator Site File. Adapted from the FDA-MHRA symposium publication
Electronic Health Records
EHRs pose regulatory and data integrity challenges due to their complexity, the absence of their direct control by sponsors, Contract Research Organizations (CROs), and investigators, and the fact that they are not designed specifically for clinical research. Unless an effective QMS is in place, the use of EHRs can also jeopardize the quality and integrity of study data, primarily for the following reasons:
- Discrepant information and types of data entered
- Transcription errors during manual data transfer to eCRFs
- Errors when using interoperable EHR and Electronic Data Capture (EDC) systems
Data loss or incomplete data collection is an additional risk arising from the lack of direct access of study monitors to EHRs and is considered one of the major data integrity challenges. According to the workshop article, it is a common practice for investigators and clinical staff to produce certified copies of EHRs and not grant direct access to EHRs to study monitors during remote or on-site monitoring visits. This issue, in combination with COVID-19-related restrictions, has been one of the topics that led to a recent update in the MHRA Guidance regarding the access to EHRs by sponsor representatives in clinical trials, published in early September 2021. The MHRA now
- Expresses its preference for monitors or auditors to have remote access to EHRs via direct system login, rather than relying on investigator site staff to use screen sharing of EHR systems
- Advises investigator site personnel to upload scanned or electronic copies of source documents into secure portals, highlighting that this constitutes direct access only if a complete and certified copy of the EHR system is provided within the portal
- Proposes for short-term mitigations, recognizing the burden and complexity of the implementation of changes to the systems
In conclusion, the article notes that both agencies are continuously adapting their ways to facilitate the use of novel clinical trial paradigms. It also emphasizes that independently of the method and technologies used for the trial conduct, maintaining data integrity and the safety of trial participants should always be the focus.
Any concerns? Reach out to us! If you need any support on dealing with the topics mentioned in this article, please reach us today for a conversation.